
Decoding Neurogenetic Answers- Case Study #1

Patient Clinical Information: The proband is an 18-month-old female presenting with developmental delays and multiple dysmorphic features. Previous testing identified two known pathogenic variants linked to acyl CoA dehydrogenase deficiency.
Family History: The proband’s mother reports a history of hypoglycemia and epilepsy since childhood, both of which are not currently seen in the proband.
Testing Ordered: MNG Exome™ Trio Sequencing + mtDNA
Genetic Findings: Exome sequencing detected a heterozygous duplication of four nucleotides in the gene SETD2. The variant was detected in 42% of the reads in the proband, which is consistent with heterozygosity. However, the variant was only detected in 14% of reads for the patient’s mother, consistent with her being mosaic for this variant.
The SETD2 gene acts in an autosomal dominant manner. This particular variant is expected to result in premature truncation of the protein, which is a known mechanism of disease for SETD2.
Several other variants were also detected, both pathogenic and VUS, which may be consistent with additional clinical presentations identified in this patient.
Outcome: The diagnosis for this patient is consistent with Luscan-Lumish syndrome. This disorder is characterized by a variety of features, including dysmorphic features and developmental delay, which is consistent with the clinical history provided for the patient. Individuals with this syndrome may also present with seizures. Since the patient’s mother is mosaic for this variant, this may explain her history of epilepsy. It is important to note that while the proband is not reported to have seizures at this time, they may be at risk for developing seizures during their childhood. In this case, the presence of mosaicism in a known pathogenic variant explains two seemingly different phenotypes in a patient and her mother.
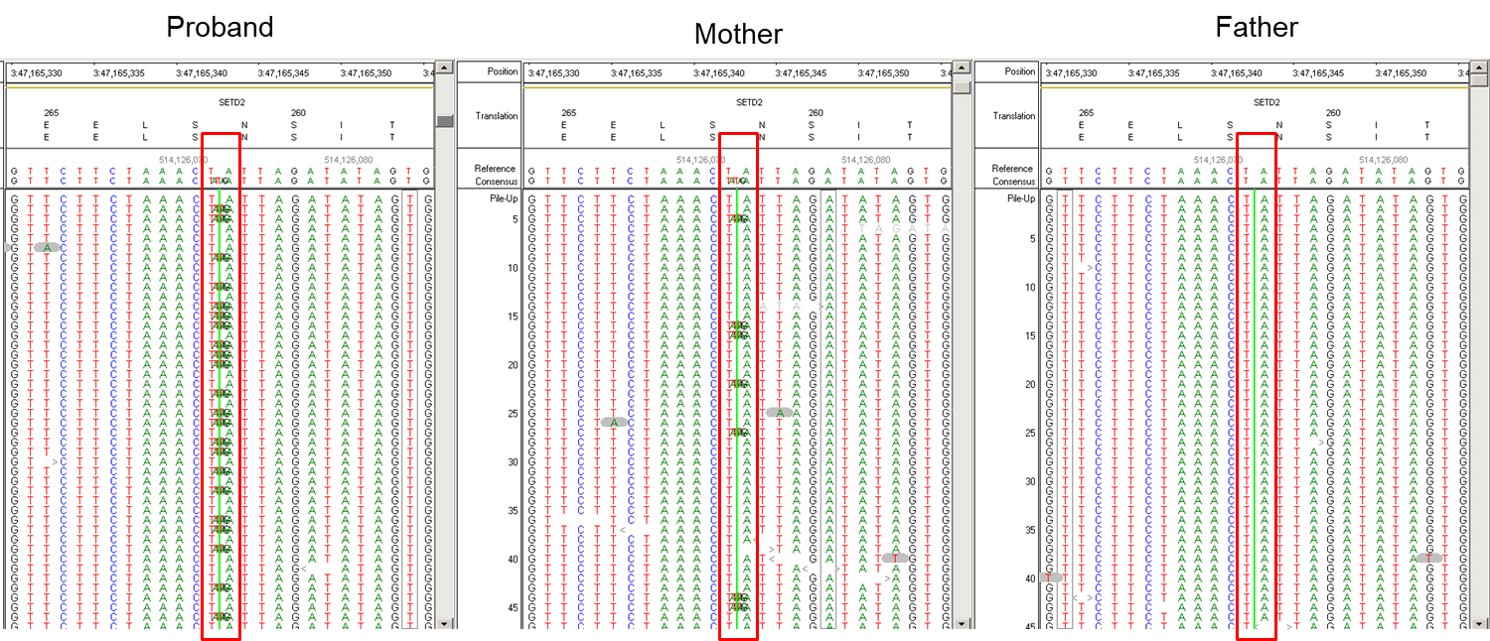
Figure 1: A heterozygous SETD2 c.779_783dup p.Ser262Leufs*4 variant (72/173 reads, 42%) was detected in the proband. The same variant was also detected at low levels (20/140 reads, 14%) in the patient’s mother, and not found in the father.
Reported by: Rebecca Kelly, MB(ASCP)CM